曾经开发出Alphago、战胜人类顶尖棋手的DeepMind团队是其中的佼佼者 , 其团队的强大和资源雄厚是一般实验室无法企及的 。 2020年12月1日 , 他们在生物领域展现出实力 , 在两年一度的权威蛋白质结构预测评估竞赛(CASP)中用AlphaFold2击败其他参赛团队 。
CASP是由马里兰大学John Moult教授等人于1994年组织 。 竞赛使用的是最新解决且尚未在蛋白质数据库(PDB)中存放或公开披露的结构 , 结构生物学家们利用X射线晶体学、核磁共振波谱学、冷冻电镜的方法 , 把这些蛋白质的结构解析出来 。 做蛋白质结构预测的团队则利用计算机程序来预测它们的结构 。 最后由独立的科学家团队则把计算机预测的模型和实验室的结构对照 , 分析不同计算机算法的预测结果 。 这是一种“双盲”测试 , 长期以来一直是评价结构预测准确性的金标准 。
去年的CASP14共有84个常规题目 , 其中有14题因为生物实验没给出确定结构等原因被取消或延缓 , 其他70个题目的单体和复合物蛋白质所含有的氨基酸个数从73到2180不等 。
19个国家的215个小组参加了CASP14 。 DeepMind公司的AlphaFold2预测的大部分结构达到了空前的准确度 , 不仅与实验方法不相上下 , 还远超解析新蛋白质结构的其他方法 。
文章图片
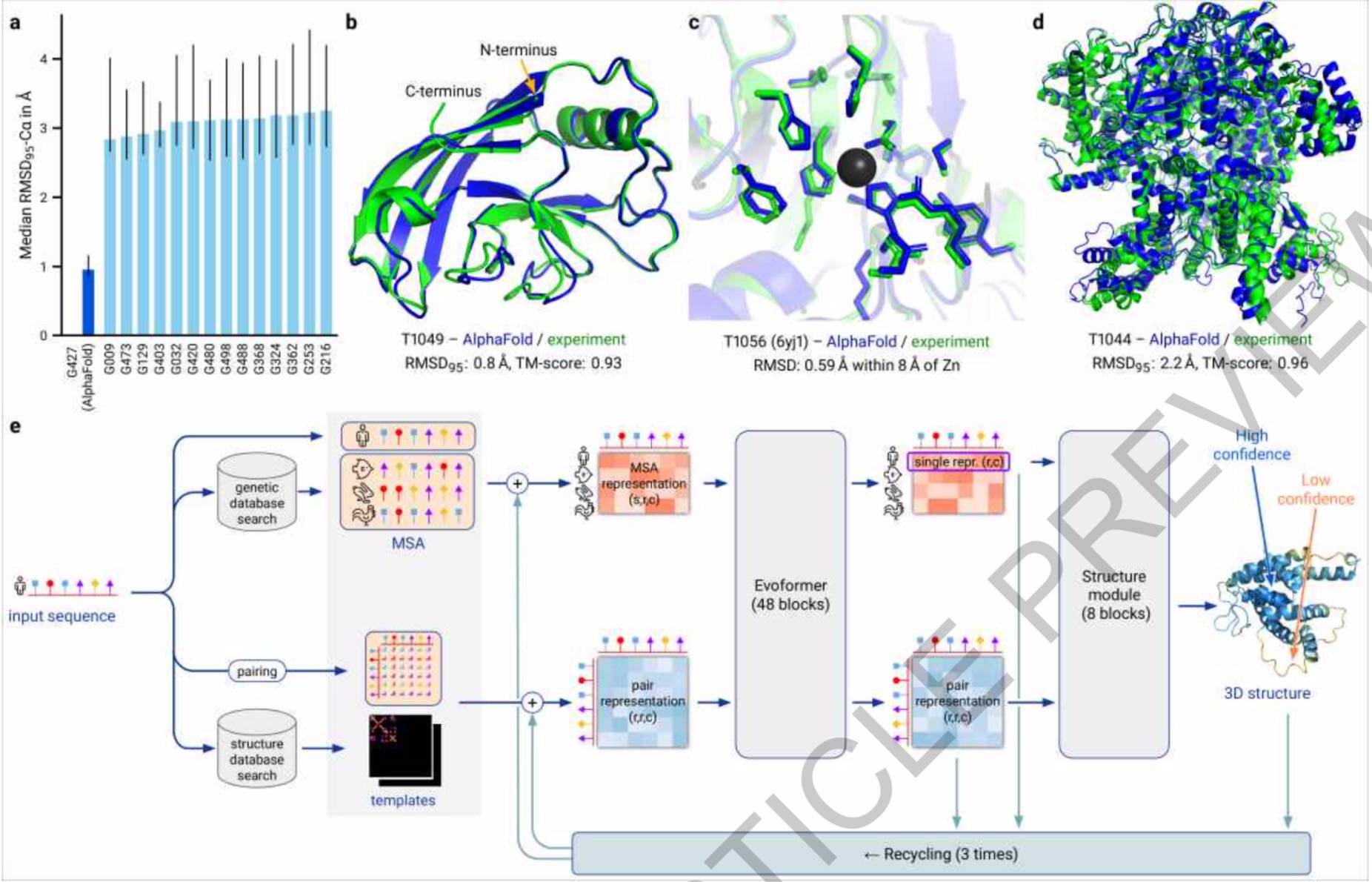
将实验方法得到的蛋白质结构叠加在AlphaFold2的结构上 , 组成蛋白质主链骨架的叠加原子之间的距离中位数(95%的覆盖率)为0.96埃(0.096纳米) 。 成绩排第二的方法只能达到2.8埃的准确度 。
AlphaFold2的神经网络能在几分钟内预测出一个典型蛋白质的结构 , 还能预测较大蛋白质(比如一个含有2180个氨基酸、无同源结构的蛋白质)的结构 。 该模型能根据每个氨基酸对其预测可靠性进行精确预估 , 方便研究人员使用其预测结果 。
AlphaFold2最终被Moult评价道 , “在某种意义上 , 问题已经解决了” 。
【科学界|蛋白质结构预测哪家强?两大国际团队同日在顶刊开源代码】值得一提的是 , 在最新发布的论文中 , DeepMind还简化了AlphaFold2 。 AlphaFold的首席研究员John Jumper说 , “这个网络需要几天的计算时间来生成CASP的一些蛋白质的结构 , 而开源版本的速度要快16倍 。 根据蛋白质的大小 , 它可以在几分钟到几小时内生成结构 。 ”
受AlphaFold2的启发 , 华盛顿大学医学院生物化学家、蛋白质设计研究所所长David Baker等人开发了RoseTTaFold 。 华盛顿大学医学院官网对该研究的介绍称 , 在高精度的蛋白质结构预测方面 , Baker等人“在很大程度上重现了DeepMind团队的表现 。 ”
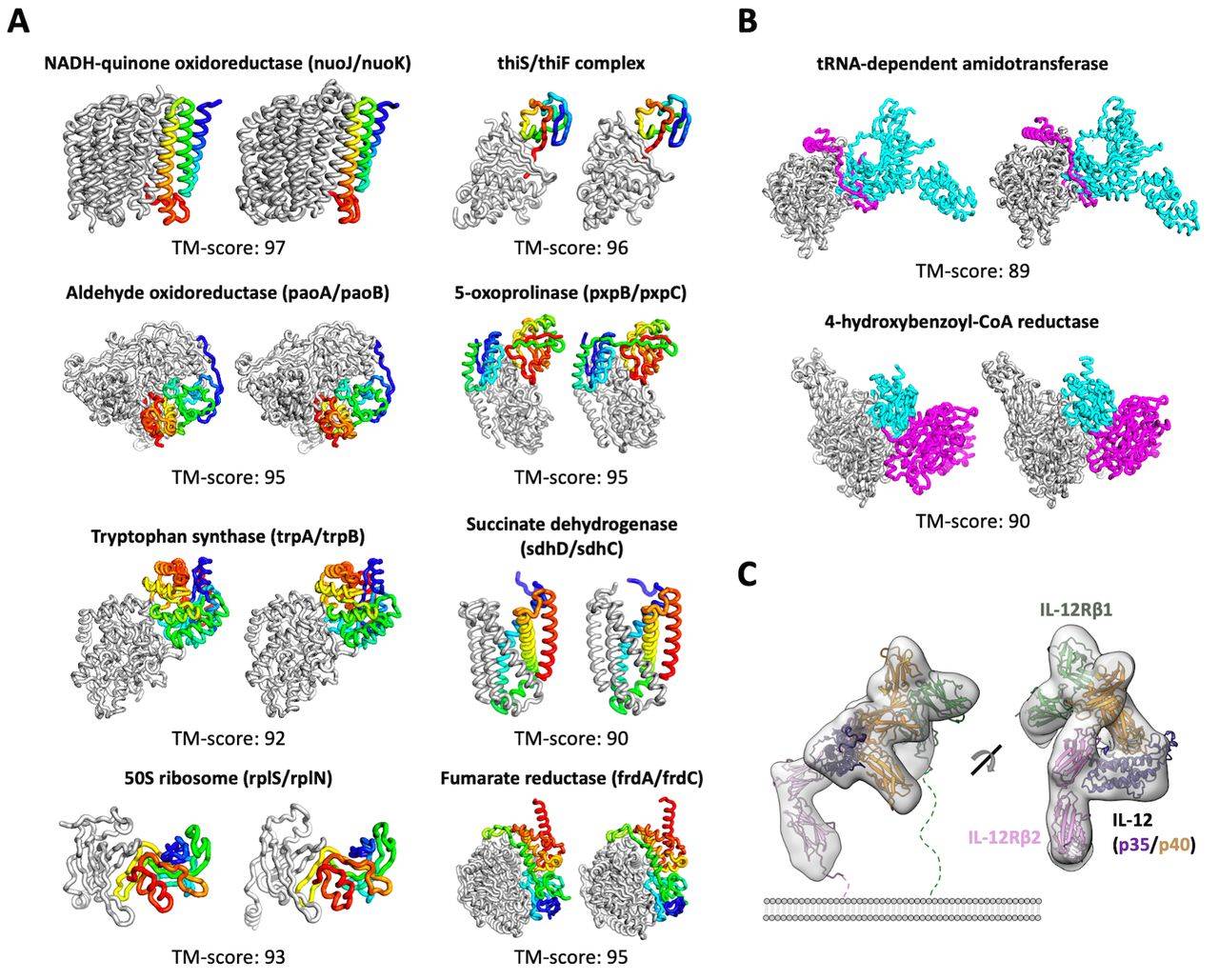
相较于AlphaFold2只解决了单个蛋白质的结构 , RoseTTaFold不仅适用于简单的蛋白质 , 也适用于蛋白质复合物 。 据介绍 , RoseTTaFold利用深度学习技术 , 根据有限信息准确、快速地预测蛋白质结构 。 从结构上来看 , RoseTTAFold 是一个三轨(three-track)神经网络 , 它可以兼顾蛋白质序列的模式、氨基酸如何相互作用以及蛋白质可能的三维结构 。 在这种结构中 , 一维、二维、三维信息来回流动 , 使得网络能够集中推理蛋白质的化学部分与它的折叠结构 。
文章图片
巴塞尔大学的计算结构生物学家Torsten Schwede对《科学》杂志说 , 许多生物功能依赖于蛋白质之间的相互作用 。 “直接从序列信息中处理蛋白质-蛋白质复合物的能力使其对生物医学研究中的许多问题极具吸引力 。 ”
Baker同时坦言 , AlphaFold2的结构更加准确 。 但是根特大学的结构生物学家Savvas Savvides说 , Bake实验室的方法更好地捕捉到了“蛋白质结构的本质和特性” , 比如识别从蛋白质侧面伸出的原子串 , 这些特征是蛋白质之间相互作用的关键 。
特别声明:本站内容均来自网友提供或互联网,仅供参考,请勿用于商业和其他非法用途。如果侵犯了您的权益请与我们联系,我们将在24小时内删除。